Table Of Content

Together with researchers from the pharmaceutical company Roche and other cooperation partners, the ETH team tested the new process and demonstrated what it is capable of. The scientists searched for molecules that interact with proteins in the PPAR class – proteins that regulate sugar and fatty acid metabolism in the body. Several diabetes drugs used today increase the activity of PPARs, which causes the cells to absorb more sugar from the blood and the blood sugar level to fall. Dr. Scott R. Lokey is a Professor of Chemistry in the Department of Chemistry and Biochemistry at the University of California, Santa Cruz. His research group focuses on the relationship between molecular structure and drug-like properties, especially cell permeability. The scientists searched for molecules that interact with proteins in the PPAR class -- proteins that regulate sugar and fatty acid metabolism in the body.
Meet the UC Campus Leads
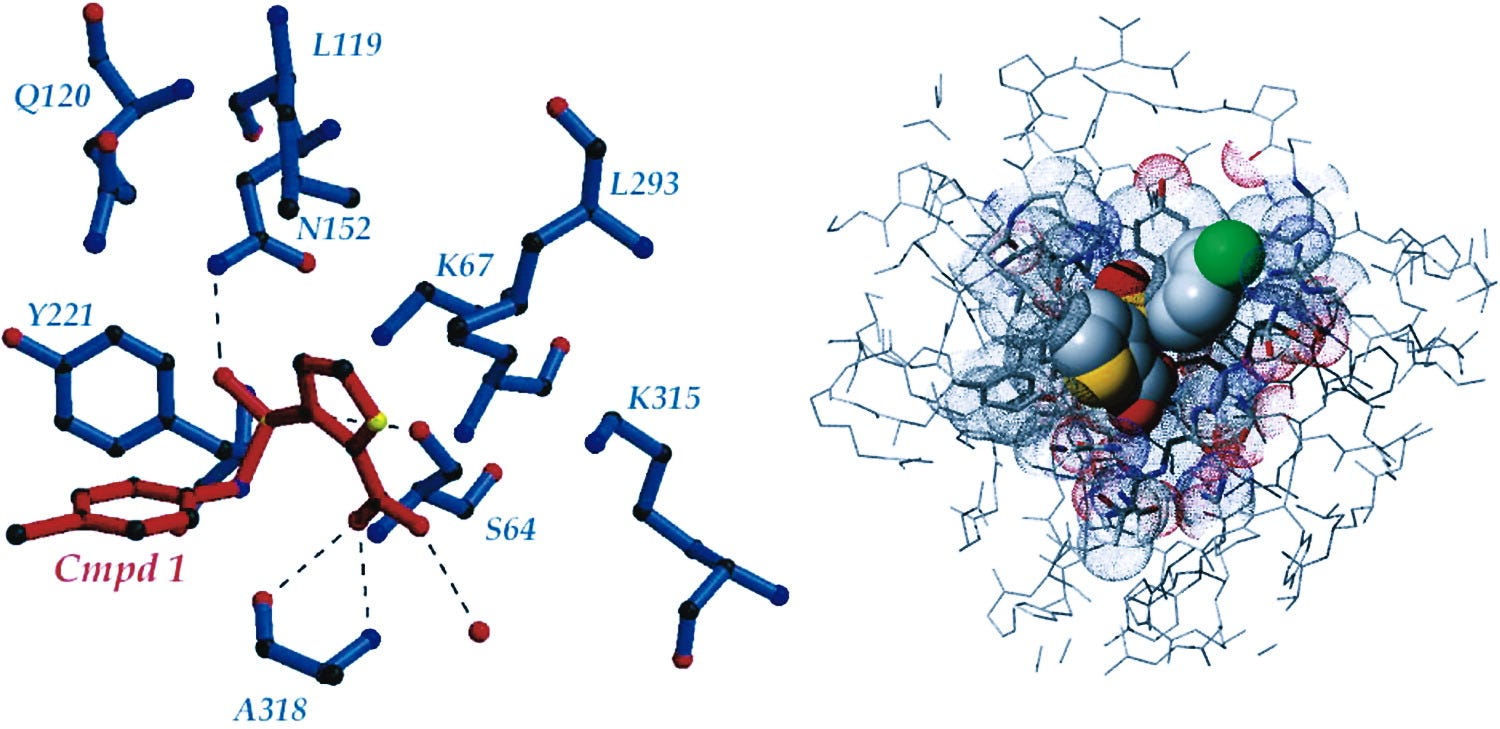
Here, among the great achievements are the techniques of molecular dynamics and molecular docking. The molecular mechanics is based on a set of empirical energy functions called a force field. The development of a drug candidate that inhibits a cancer-related protein was stopped due to insufficient selectivity. The X-ray data of the complex of the molecule with the protein concerned is available. This is a possible starting point for a receptor-based project aiming at the discovery of potent and selective molecules.
Sequence-based drug design as a concept in computational drug design
A molecule can be designed that has optimal interactions with the 3D structure of the target protein. For example, biological activity can be expressed quantitatively as the concentration of a substance required to give a certain biological response. Additionally, when physicochemical properties or structures are expressed by numbers, one can find a mathematical relationship, or quantitative structure-activity relationship, between the two.
Repositioning proton pump inhibitors as anticancer drugs by targeting ARF1
In this series, we use specific examples to show how ETH is working on joint projects with industry, NGOs and the authorities to harness AI for Switzerland, thereby creating added value for our country. Based on that, it designs molecules that bind specifically to the protein according to the lock-and-key principle so they can interact with it. A new computer process developed by chemists at ETH Zurich makes it possible to generate active pharmaceutical ingredients quickly and easily based on a protein’s three-dimensional surface. Melanie Cocco has a PhD in Organic Chemistry from Penn State and was an NIH postdoctoral fellow at Yale in the Department of Biophysics and Biochemistry.
Treatment for Designer Drugs
How AI is Shaping Drug Discovery at AION Labs - Healthcare IT Today
How AI is Shaping Drug Discovery at AION Labs.
Posted: Thu, 25 Apr 2024 15:00:58 GMT [source]
The desired region for the generated molecules, which satisfies both, novelty and the predicted bioactivity requirements, is highlighted by a blue box located in the upper right corner of the plot. The ranking criteria were PPARγ + PPARδ dual-target affinity and structural novelty (left, 1 & 6–9), or PPARγ single-target affinity and structural novelty (right, 2, 6 & 10–12). C Examples of non-carboxylic head groups and secondary amides from the top-100 molecules ranked for PPARγ, where the gray shaded R group represents an aromatic moiety connected to a linker. Several deep learning models have been proposed to use protein sequences as input19,20,21,22,23,24,25,26,27,28. However, none have thoroughly verified the concept of the sequence-to-drug paradigm. First, we designed TransformerCPI2.0 as a fundamental tool of the sequence-to-drug paradigm, which exhibited generalization ability across proteins and chemical space.
CT26 cells transplanted tumor growth
GQSAR also considers cross-terms fragment descriptors, which could be helpful in identification of key fragment interactions in determining variation of activity. In this context FB-QSAR proves to be a promising strategy for fragment library design and in fragment-to-lead identification endeavours. Numerous drug targets exhibit multiple binding sites, including orthosteric sites and various allosteric sites94.
Artificial intelligence is taking over drug development - The Economist
Artificial intelligence is taking over drug development.
Posted: Wed, 27 Mar 2024 07:00:00 GMT [source]
The history of the Thomas J. Long School of Pharmacy and Health Sciences at the University of the Pacific goes back to February 1955, when the first class was held. After years of growth and dedication, the school now offers numerous graduate and professional degrees in areas such as speech-language pathology and audiology, pre-health science, and pharmacy. The Pharmaceutical and Chemical Sciences program is available as a Master of Science, Doctor of Philosophy, or a dual Doctor of Pharmacy/Doctor of Philosophy.
Our findings indicated that Ocotillone and Subsessiline have potential antileishmanial effects at pH 5 and 7, respectively, due to their high binding affinity to MOGS and interactions in the active center. Furthermore, these compounds were non-toxic and had the potential to be administered orally. This research indicates the promising anti-leishmanial activity of Ocotillone and Subsessiline, suggesting further validation through in vitro and in vivo experiments. By leveraging an interactome-based deep learning approach and employing a graph-to-sequence neural network architecture, DRAGONFLY addresses certain challenges commonly encountered in generative molecular design methods. Its ability to combine structure-based and ligand-based approaches, as well as its capacity to incorporate desired properties makes it a potentially useful tool for medicinal chemistry. The central component of DRAGONFLY is its drug-target interactome, which captures the connections between small-molecule ligands and their macromolecular targets.
Once trained, the CLM can generate virtual molecular libraries tailored to the specific task at hand11. Some CLM approaches integrate reinforcement learning techniques, enabling an additional level of fine-tuning to optimize the properties of the generated molecules12,13. After discovering inhibitors for SPOP, we applied this concept to discover hits for a more challenging target RNF130 whose crystal structure is unknown.
The magnetic beads were then washed 3 times with PBST, and the immunoprecipitated proteins were eluted with 1 × SDS loading buffer at 100 °C for 5 min. Second, we filtered pan assay interference compounds (PAINS) and clustered these molecules automatically based on their extended-connectivity fingerprints (ECFP), obtaining approximately 800 clusters. Third, we filtered these compounds by the Lipinski rules and selected representative compounds from top ranked clusters. Finally, a total of 82 candidates were purchased for further experimental evaluation.
Celltiter-glo (CTG) reagent (G7572, Promega) was prepared according to the manufacturer’s instructions. The plates were then shaken for 2 min and incubated for an additional 15 min at room temperature. NMR spectroscopy experiments were performed using a 600 MHz spectrometer (AVANCE III, Bruker) to validate protein–ligand interactions. In Carr-Purcell-Meiboom-Gill (CPMG) and saturation transfer difference (STD) NMR experiments, compound was dissolved to a final concentration of 200 μM in a solution of PBS formed with D2O containing 5 μM SPOPMATH protein and 5% DMSO‑d6. All the baseline models, including TransformerCPI, CPI-GNN, GraphDTA(GAT-GCN), MolTrans and GCN, were trained on the ChEMBL dataset with their own hyperparameters. Only the learning rate, weight decay rate and dropout rate were subjected to a hyperparameter search.
On the other hand, DeepMGT-DTI method [19] converted drug SMILES into the molecular graphs and utilized molecular complementary graph convolutional neural networks (MCGCN) and Transformers to extract high-level features from drug molecular structures. Additionally, it employed CNN to extract high-level features from target sequences. By concatenating the extracted features, the FC network predicted the presence or absence of drug-target interaction. Another approach, described in reference [20], involved using two graph convolutional networks (GCNs) to extract structural features.
The funder had no role in the design of this review; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish it. Based on the reasons described above, it is obvious that drugs are high-value-added products and the new drugs are highly expensive. Unfortunately, even nowadays drugs are non-affordable products for most people on this planet. This last phase is known as a post-marketing surveillance and it is practically endless, it continues until the drug is on the market. The FHE Health team is committed to providing accurate information that adheres to the highest standards of writing. If one of our articles is marked with a ‘reviewed for accuracy and expertise’ badge, it indicates that one or more members of our team of doctors and clinicians have reviewed the article further to ensure accuracy.








No comments:
Post a Comment